विश्व थैलेसीमिया दिवस मनाना
भारत में, हर साल लगभग 12,000 बच्चे आनुवंशिक रक्त विकार के साथ पैदा होते हैं जिससे स्वास्थ्य संबंधी गंभीर जटिलताएँ होती हैं। थैलेसीमिया - सबसे आम आनुवंशिक रक्त विकारों में से एक - एक आनुवंशिक स्थिति है जिसमें हीमोग्लोबिन के असामान्य उत्पादन के परिणामस्वरूप स्वास्थ्य संबंधी चिंताएँ अलग-अलग स्तर की होती हैं। भारत में, लगभग 25 में से 1 भारतीय बीटा-थैलेसीमिया का वाहक है।
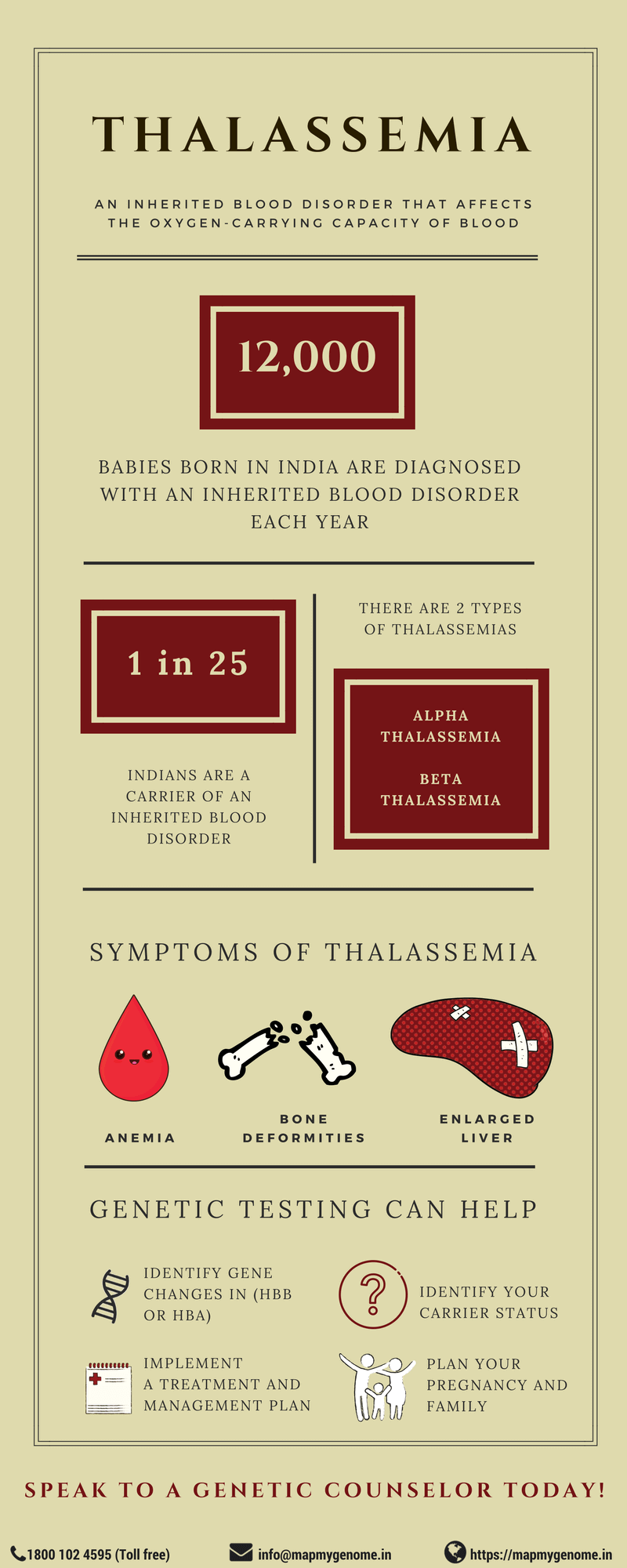
अल्फा थैलेसीमिया
हीमोग्लोबिन की असामान्य रूप से बनी अल्फा श्रृंखलाएं अल्फा थैलेसीमिया का कारण बनती हैं। इसे चिकित्सकीय रूप से दो रूपों में वर्गीकृत किया गया है:
हाइड्रोप्स फेटालिस / एचबी बार्ट सिंड्रोम (अल्फा थैलेसीमिया मेजर) — एक गंभीर रूप; बच्चे मृत पैदा हो सकते हैं या जन्म के तुरंत बाद मर सकते हैं। गर्भवती माताओं को भी उच्च रक्तचाप, समय से पहले प्रसव और असामान्य रक्तस्राव सहित जटिलताओं का खतरा हो सकता है
एचबीएच रोग — एक हल्का रूप जो शैशवावस्था या प्रारंभिक बचपन में हल्के से मध्यम एनीमिया, प्लीहा और यकृत के बढ़ने, और आंखों और त्वचा के पीले पड़ने के साथ प्रस्तुत होता है
बीटा थैलेसीमिया
बीटा थैलेसीमिया की विशेषता कम मात्रा में बीटा-हीमोग्लोबिन श्रृंखलाएं हैं जो हाइपोक्रोमिक एनीमिया का कारण बनती हैं। इसे इसमें वर्गीकृत किया गया है:
थैलेसीमिया मेजर (कूली का एनीमिया) — नैदानिक लक्षण 6 से 24 महीनों के बीच दिखाई देते हैं; हर 2-3 सप्ताह में नियमित रक्त आधान की आवश्यकता होती है
थैलेसीमिया इंटरमीडिएट — प्रारंभिक या देर से बचपन में लक्षण दिखाई देते हैं; मध्यम से गंभीर हड्डी की विकृति और यकृत/प्लीहा का बढ़ना
आनुवंशिकी और वंशानुक्रम
अल्फा थैलेसीमिया HBA1 और HBA2 जीनों (ऑटोसोमल रिसेसिव) में विलोपन के कारण होता है। बीटा थैलेसीमिया HBB जीन (ऑटोसोमल रिसेसिव) में रोग-उत्पादक परिवर्तनों के कारण होता है। जब दोनों माता-पिता वाहक होते हैं, तो उनके बच्चों में थैलेसीमिया होने की 25% संभावना होती है, वाहक होने की 50% संभावना होती है, और अप्रभावित होने की 25% संभावना होती है।
थैलेसीमिया के लिए कैरियर स्क्रीनिंग
भारतीय वंश के किसी भी जोड़े के लिए गर्भावस्था की योजना बनाने, थैलेसीमिया के पारिवारिक इतिहास वाले व्यक्तियों और दाता शुक्राणु या अंडे का विकल्प चुनने वाले व्यक्तियों के लिए वाहक स्क्रीनिंग की सिफारिश की जाती है। जब दोनों माता-पिता वाहक होते हैं, तो उनके बच्चों को थैलेसीमिया के कारण जटिलताओं का खतरा होता है।
थैलेसीमिया वाले परिवारों के लिए संसाधन
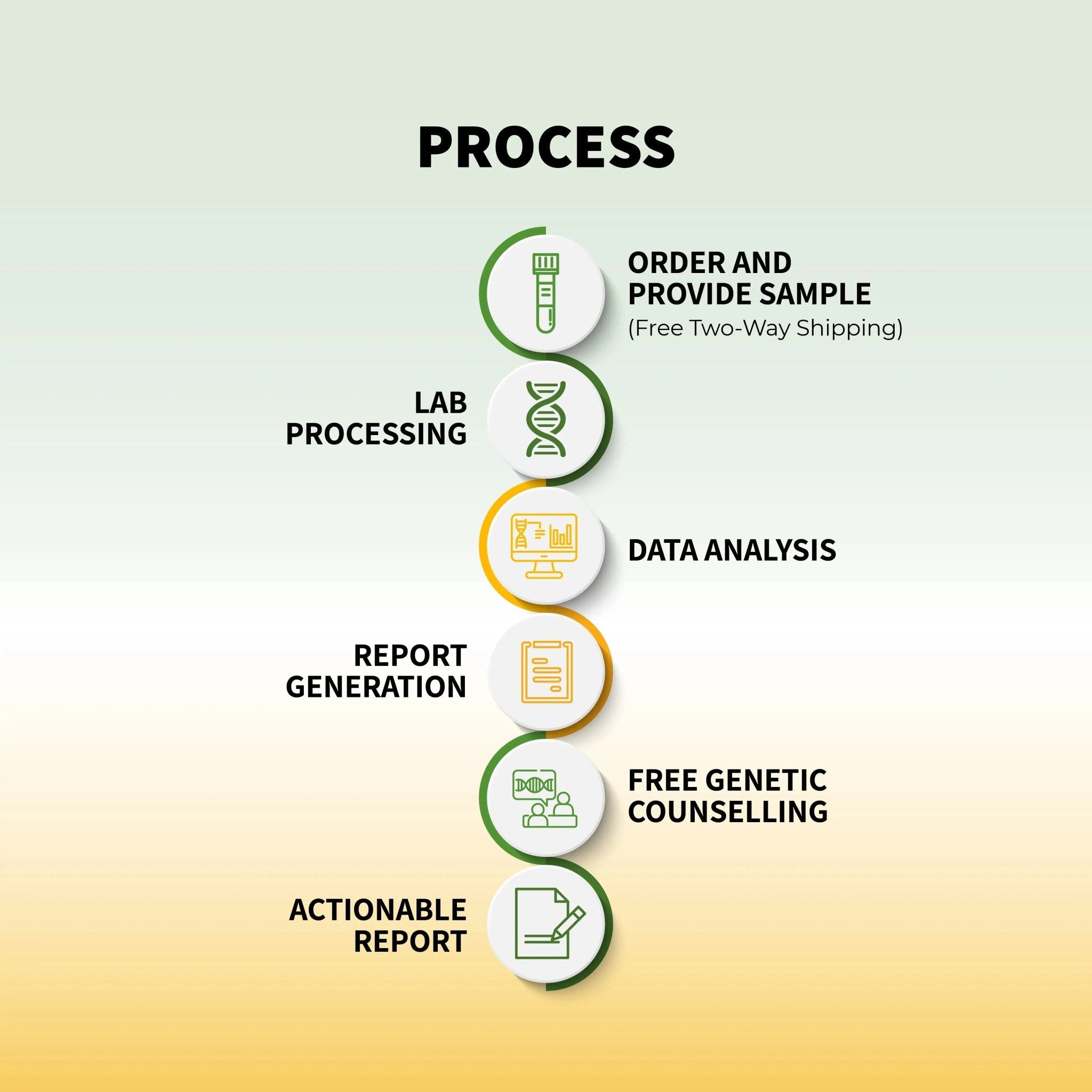
थैलेसीमिया के लिए कैरियर स्क्रीनिंग और जेनेटिक काउंसलिंग
मैपमायजीनोम भारत की सबसे अनुभवी टीम से प्रमाणित जेनेटिक काउंसलिंग के साथ 171 आनुवंशिक स्थितियों - जिसमें अल्फा और बीटा थैलेसीमिया शामिल हैं - को कवर करते हुए कैरियर स्क्रीनिंग प्रदान करता है। CAP & NABL-मान्यता प्राप्त प्रयोगशाला।
कैरियर स्क्रीनिंग का अन्वेषण करें → जेनेटिक काउंसलिंग बुक करें →